Sanofi completes buyout of Vigil Neurosciences for a small molecule TREM2 agonist program
August 14th, 2025 By John Widen
Sanofi completed a buyout of Vigil Neurosciences this past week that was announced in May
of this year for 8$/share or approximately $470 million. Previously,
Sanofi invested $40 million in Vigil last June (2024). This deal closed around the same time that Vigil's
TREM2 (triggering receptor expressed on myeloid cells 2) antibody agonist,
VG101 (iluzanebart), failed to meet biomarker and clinical endpoints for a phase 2 trial in a rare
neurological disease, CSF1R-related Axonal Spheroids and Pigmented Glia (CSF1R-ALSP).
Iluzanebart was meant to address a subpopulation of patients with loss-of-function mutations
in CSF1R (Colony Stimulating Factor 1 Receptor) causing ALSP. TREM2 signaling overlaps with the
CSF1R downstream pathway involving phosphoinositide-3-kinase (PI3K) and modulates
CSF1R cell surface expression. After the phase 2 failure, iluzanebart was dropped from the
Sanofi deal and given to Amgen, leaving only the TREM2 small molecule agonist
program that completed a phase 1 trial in February of 2025. I thought this would be a good
time to focus on TREM2 as a drug target for Alzheimer’s disease, cover the recent clinical
failures of TREM2 antibody agonists, and discuss some of the TREM2 agonist chemical matter
in the literature.
Vigil Neurosciences is a single target biotech company focused on a cell surface receptor TREM2,
which is a drug target for Alzheimer's disease because it is a genetic risk factor identified in
large population genome-wide association studies (GWAS)
and has biological relevance to neurodegeneration.
The founder’s of Vigil spun out the TREM2 assets from Amgen (hence, giving iluzanebart back) and raised about $140 million
in a series A and B funding round. TREM2 is a lipid sensing receptor expressed predominantly in microglia.
Upon binding of extracellular lipids and debris, TREM2 activates a signaling cascade through phosphorylation
of SYK (Spleen associated tyrosine kinase) via an adapter protein DAP12 (DNAX-activating protein 12, a.k.a TYROBP).
Downstream signaling results in increased phagocytosis, mobility, and lysosomal activity.
The therapeutic approach for TREM2 is to agonize the receptor to induce these desirable
effects in microglia for the elimination of extracellular lipids and debris (e.g. Aβ plaques).
If you want to learn more about TREM2 biology,
I would suggest this
review to get started.
Amgen and Vigil have published 10 total patent applications focused on small molecule TREM2 agonists.
Based on timing, the clinical candidate, VG-3927, is likely from the first two patent applications published in 2021.
I am not going to go through the chemical matter from these patent applications in this article, but feel free to take a look
for yourself with the list of WO patent application IDs below.
- WO2025128873
- WO2025128848
- WO2024233847
- WO2024233848
- WO2023086801
- WO2023086799
- WO2023086800
- WO2022236272
- WO2021226135
- WO2021226629
In uncanny timing, there was a
report published in ACS Med. Chem. Lett.
this past week describing a small molecule TREM2 agonist C1,
a structurally related analogue of Vigil’s alleged clinical candidate VG-3927
(Fig. 1). (NOTE: The article is behind a paywall.
So, my apologies if you do not have access.)
I say alleged, because this is the first time to my knowledge that the structure of VG-3927 has been disclosed.
I haven’t attended very recent presentations from Vigil, but I cannot find any information suggesting
that they had a first-time disclosure. Regardless, the publication is a little odd because
it throws VG-3927 under the bus a little bit.
The authors suggests that VG-3927 does not activate TREM2 in a dose dependent manner in an
in vitro assay compared to the close analogue C1.
They also demonstrate that VG-3927 does not have measurable binding in an MST
assay compared to C1 that has a very weak KD of 71 µM.
In addition, the authors describe that C1 has superior pharmacokinetic
parameters than VG-3927. Looking at the in vitro DMPK data presented,
I have a hard time agreeing that the PK parameters are any different between the two
molecules (Fig. 1). Without in vivo PK data to verify that
these two molecules are differentiated in terms of half-life, clearance (CL), brain
penetration (Kp,u,u, Bu/Pu), or any safety parameters,
I think these claims are unfounded and surprised that this article passed through the
peer review process without dialing back the interpretation of the results.
These data presented in the ACS Med. Chem. Lett. publication go against a swath of
data disclosed by Vigil in their presentations. I cannot show screen shots from Vigil’s
presentations without permission. However, if you look on their
website,
they have quite a bit of data suggesting direct modulation of TREM2 signaling
including in vitro assays measuring pSYK, which is phosphorylated in a
TREM2-dependent, dose response manner. Additionally, Vigil demonstrates that VG-3927 has a
TREM2 clustering effect by Western blot (WB) that has been
described as a
mechanism of activation by Denali Therapeutics. Additionally,
TREM2-dependent agonism by VG-3927 increases relevant cytokine expression
by RNAseq such as Cxcl10 and Ccl2. Albeit the cytokine activation is relatively
weak (2-3 fold change from baseline). I will say that Vigil’s pre-clinical data
demonstrating direct interaction with TREM2 is weak overall, but these data demonstrate
that there is TREM2-dependent activation by VG-3927.
Phase 1 biomarker data presented by Vigil also suggests direct TREM2 modulation
as measured by soluble TREM2 (sTREM2) in CSF (cerebral spinal fluid) of patients dosed
with the drug. Upon agonism of TREM2, the receptor is internalized and unavailable
for ‘shedding’,
hence an expected reduction in sTREM2 in the CSF. It is interesting
to note that Vigil was measuring other downstream biomarkers in the Phase 1 trial such as CSF1R.
However, Vigil did not include that analysis in their presentations of VG-3927
Phase 1 clinical data,
likely because there was no modulation.
So, to-date Vigil (now Sanofi) has not published any evidence of downstream TREM2 pathway
activation in humans other than sTREM2 levels, which is only indicative of direct target engagement.
The same goes for the TREM2 agonizing antibody, iluzanebart. Vigil claims that a small molecule
approach to agonizing TREM2 is superior to an antibody because the sTREM2 in CSF binds to the
antibodies and restricts activation of cell-surface TREM2 whereas
small molecules only bind to surface expressed TREM2. Vigil disclose data in support of this
comparing VG-3927 to Alector’s TREM2 antibody agonist AL002.
Speaking of other TREM2 agonists, the Vigil-Sanofi deal is in the wake of two other clinical
failures of TREM2 antibody agonists. Alector’s AL002
did not meet any endpoints in a
Phase 2 clinical trial to treat Alzheimer's disease (NCT04592874).
AL002 was dosed alone every four weeks in a randomized, double blind, placebo controlled study.
The TREM2 antibody agonist did not improve cognitive scores, reduce Aβ levels measured by PET imaging,
nor did it modulate any measured biomarkers. The biomarkers measured from the Phase 2 trial have not been disclosed.
Counter to Vigil’s small molecule VG-3927, AL002 did modulate sCSF1R levels in a Phase 1 trial.
In addition to the lack of therapeutic effect, amyloid-related imaging abnormalities (ARIA) and
infusion-related reactions were also observed during treatment.
The second clinical failure for a TREM2 antibody came from a Denali Therapeutics and
Takeda partnership in April 2023 (NCT05450549). The TREM2 antibody agonist developed by
Denali, DNL919, demonstrated much stronger TREM2 agonist effects in pre-clinical models
compared to AL002, VG101 (iluzanebart), and VG-3927. However, in a Phase 1 trial evaluating safety
DNL919 caused hematological effects (e.g. reticulocyte loss) likely due to the mechanism used by
Denali and others to actively transport antibodies past the blood-brain barrier.
This technology uses transferrin receptor (TfR) to bind to an antibody containing a
TfR recognition sequence. Upon binding to TfR present on the endothelial layer in brain vasculature
(a.k.a. blood-brain barrier), the antibody is endocytosed and can traverse into the brain parenchyma.
Antibodies containing a TfR binding element leads to an increase in antibody levels in the brain
compared to conventional antibodies. Unfortunately, TfR is also highly expressed in other tissues
including reticulocytes. This is the same technology that the Aβ clearing antibody Trontinemab
developed by Roche uses to increase antibody concentrations in the brain.
Trontinemab recently completed a Phase 1b/2 clinical trial demonstrating safety (NCT04639050).
Denali has demonstrated safety windows for other antibodies containing a TfR binding element,
but this known risk limited the therapeutic window for DNL919 and caused Denali and Takeda to
stop the Phase 1 clinical trial. It is unclear if Denali is going to take another shot with a
different TREM2 antibody agonist at this point. Unfortunately, because DNL919 did not make it
past initial safety evaluation there is no way to tell if it would have
differentiated from the competition.
With these three clinical failures for TREM2 antibody agonists and a lack of evidence for
downstream microglia activation in these human clinical trials, the
future is quesitonable for targeting TREM2 agonism to treat rare neurological diseases or Alzheimer's disease.
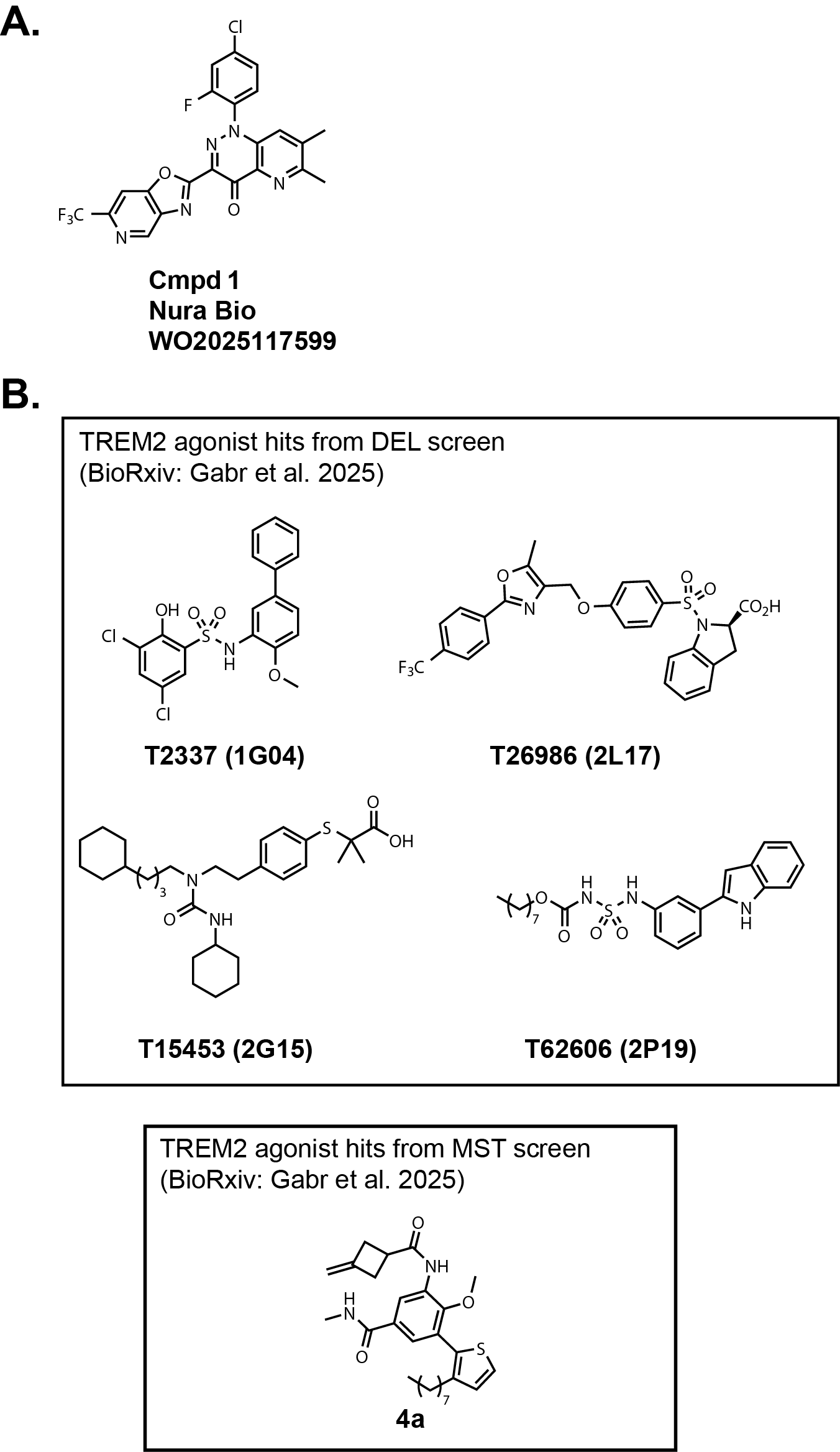
Nura Bio, who I discussed in the context of SARM1 last week, recently published a patent application
focused on small molecule TREM2 agonists (Fig. 2A, WO2025117599). It is pretty obvious that they are patent busting
Amgen/Vigil Neuroscience's original chemical matter.
In addition, two BioRxiv papers (linked
here and
here) were published recently by the same group that authored the
ACS Med. Chem. Let. paper mentioned above.
These two publications report the identification of small molecule hits from a DEL
(DNA-encoded library) screen and a small library screen using an MST-based assay (Fig. 2B).
Otherwise, the literature is pretty scant outside of these publications in terms of small molecule modulators
of TREM2. Undoubtedly, other companies have started drug discovery programs focused on this target.
Time will tell if any of those efforts amount to anything.
I am speculating that Sanofi is interested in TREM2 agonists to combine with other
Alzheimer’s therapies such as Aβ targeting antibodies. Maybe Sanofi already has internal
data to suggest that there is a path forward for combination therapies focused on TREM2 and Aβ.
In theory, these two pathways could act synergistically to clear extracellular debris and plaques
in Alzheimer’s disease. There is certainly plenty of pre-clinical data in support of this hypothesis.
Given the lack of biomarker movement in humans for VG-3927 in the context
of the recent TREM2 antibody clinical failures, it would be interesting to know what data excited
Sanofi to the point of buying out Vigil Neurosciences for $470 million. I'll stop there, thanks for reading.
The site does not have a comments section yet! Hopefully, very soon! Until then please drop me a line at jwiden@chemjam.com.
If you provide comments on my articles I reserve the right to post them on this website as additional commentary. My goal is to have an open discussion!