Part 3: Patent Busting A Literature-based Approach
August 27th, 2025 By John Widen
Welcome back! This is Part 3 of the Patent Busting Literature-based Approach blog series!
Part One
of this series focuses on searching for patent applications to efficiently identify
relevant chemical matter for a protein target of interest. Finding relevant patent
applications can be a daunting task whether the focus is on a very popular target like KRAS or a
target that has very limited public chemical matter. Part One has tips to help narrow down the search.
Part two of this blog series covers the various sections of a patent application and
what to look for when evaluating the chemical matter and protein target of interest. The claims, definitions,
and exemplified molecules are the most important to identify ways to differentiate from prior art.
However, there are many other aspects of patent
applications that are helpful including the background, assay information and data, and prior art.
I encourage everyone interested in how to go about a literature-based/information-driven
approach to go back and read through Part One and Two of this blog series if you missed it!
Part three will focus on identifying the GAPS in the claims. To do this, I will continue focusing on a STING
(stimulator of interferon genes) antagonist
patent application from Boehringer Ingelheim, WO2024089155.
This is the most exciting part of patent busting in my humble opinion.
There are GAPS in claims because it would take a significant amount of time and
resources to synthesize every possible analogue within a chemical series.
As I mentioned previously, the claims are limited by the compounds exemplified
and the prior art (i.e. other patent applications). The goal of a patent application
is to destroy novelty and grab as much chemical space as possible to protect intellectual property.
This is why claims exist in the first place. However, it is quite an impossibility to
cover the chemical space so broadly that other folks cannot differentiate outside of
the claims within a patent application. That is the case most of the time.
Sometimes, with a popular target and a crowded IP space, medicinal chemists can get
very creative designing novel chemical matter, making it very challenging to
identify ways to differentiate from the prior art.
As I go through the claims, I’m going to mention possibilities but not
every single last one. I'm also not going to go all the way through the claims down to Rinfinity, but I will
go through the main R groups and discuss ways to differentiate.
Certain apparent GAPS may be limited by other prior art.
Additionally, even though GAPS exist, they might be there because they would lead to
dead ends in terms of potency or ADME properties. But, if that can’t be determined by
the assay data provided in the patent application, there is really only one way to
find out: Try it out! There are other considerations such as modeling and comparing
low energy 3D conformations to exemplified molecules.
There are many structures of STING with bound molecules to the cGAMP (cyclic GMP-AMP) binding site
in the PDB database. Very briefly, a protein called cGAS (cGAMP synthase) can bind to cytosolic DNA, produce cGAMP, which binds to and activates STING.
The STING pathway is part of the innate immune response and has relevance as a drug target because of its role in
cancer and other immune related diseases. I'm not going to go into any more detail but feel free to read the introduction in the
patent application or other literature if you're interested.
Some agonists and
antagonists in the literature bind as
dimers within the large cGAMP pocket
(See PDB: 6MXE for antagonist and PDB: 6XNP for agonist bound as dimer).
It is a possibility that the antagonists in this patent application bind within the cGAMP binding site as dimers,
but it isn’t guaranteed. Binding as a dimer would definitely affect the SAR and what
types of substitutions can be made. However, I’m not going to do any computational work
for this exercise but will likely mention the possibility of certain substitutions or
changes that would likely affect these things. It is good to always consider what is known
about the binding mode of the chemical matter under evaluation and is a great head start
if there are public structural data for the patent molecules or a molecule within that series
bound to the protein target. If not, and there is a chance of success to obtain a structure,
it can certainly accelerate a drug discovery program. Highly recommended if it is a possibility!
To stay organized, I use Excel to divide up the claims
into cells with respective columns that contain notes and potential GAPS.
It is really just note taking but I think it is important to stay organized because
presenting this information to others will inevitably require going back through the claims and GAPS.
The excel file I generated from WO2024089155 is in Table 1.
I did not beautify it very much, but just wanted to provide an example of my method.
I realize everyone thinks differently. So, if there is a preferred method by all means,
but if you’re just getting into the concept of patent busting a
literature-based approach maybe these notes will be helpful.
The Notes column has references to specific exemplified molecules within
the patent application and associated activity. The first activity given is from an HTRF binding assay, the second number is
from a differential scanning fluorimetry (DSF) assay (in Kelvin, K), and the last activity value is from a whole blood assay.
The last assay is the most important in terms of potency. However, this assay is the only
one reported in this patent application where potency can be affected by protein binding.
That makes the values more difficult to compare without measuring or calculating protein binding.
There likely are not going to be >10 fold differences in free fraction for molecules that are similar, but it is still something
to keep in mind when comparing potencies. The GAPS column are potential points of differentiation that were not
included in the claims. These are not comprehensive, but again just giving an idea of my process.
One important goal when taking a literature-based approach is to differentiate from prior art.
This means that the chemical matter generated has to be outside of the claims of any published patent application.
One approach could be to simply move a nitrogen there or remove a nitrogen there and BAM(!) new chemical matter.
This has been a successful approach for ‘fast followers’ but often times it is a better approach to get to two,
three, or even more points of differentiation from the prior art. Published patent applications represent the work
of others approximately 1.5 years prior. After a group synthesizes and tests molecules in their assay(s), and
they think there is a path forward to the clinic, or at the very least think the chemical series is valuable enough,
they will put a patent application together and file it to their respective patent offices. That starts a timeline to the
patent application being published. The group can add to the patent application over the next year in the
form of exemplified molecules. At that year mark the application must be finalized
and will be published six months from that date.
There are other details about this process but I’m not going to open that can of worms here.
The point I am trying to make is that the group that filed the patent application has had a 1.5 year head start.
After that initial patent application there are likely other patent applications that will expand
on the chemical matter of the first. They will address their own GAPS with additional patent filings.
It isn’t always the case, but that likelihood is often high. The group that filed likely kept working on the project,
generated additional novel chemical matter, and filed additional patent applications because they want to
protect their IP and block others from working in the same space. It is a big game of cat and mouse.
So, it is best to differentiate from the chemical matter as much as possible to avoid getting scooped
(i.e. finding out later that the chemical matter you generated is actually covered by an application
that is yet to be published). There are other nuances of course within this game but I think that
is sufficient information for now to set up going through the claims to identify
points of differentiation (GAPS!). The take home message is differentiate as much as possible.
And in that process discover better molecules that are more potent with better DMPK/ADME properties
and/or different mechanism of modulation.
Okay. With that, let’s get into it!
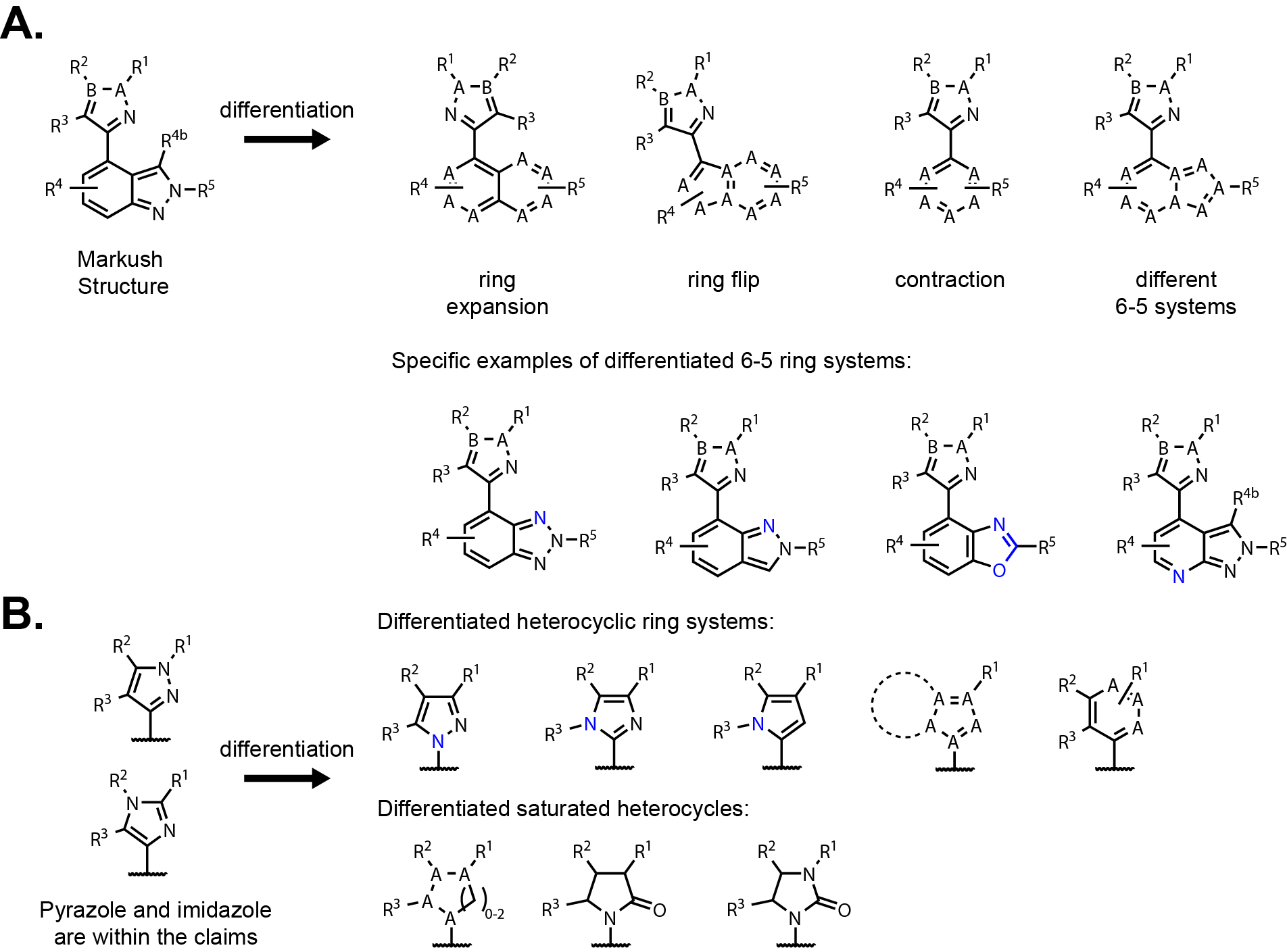
The Markush Structure:
- B-A is =C-N- or -N-C=; this means that A is C or N; B is C or N; but A and B are not N at the same time;
Starting out with the Markush structure there are already possibilities of differentiation
(Fig. 1A). There are opportunities to expand and flip the central indazole.
There are other 6-5 heteroaryls to try including benzotriazole, azaindazole, benzoxazole,
benzothiazole, bridging nitrogen heterocycles, etc. These changes to the 6-5 ring system depend
on if polarity (and lone pairs) are tolerated at various positions. It very well could be that
these were not included in the claims because some of these options were tried and lacked activity.
But again, you don’t know until you try. Keeping the 6-5 system is a safer bet
but flipping the core or expanding to a naphthalene, isoquinoline, or quinazoline-like core could be
synthetically accessible and simple to try. A simple analogue to evaluate the possibility might lead to a new series.
Comparing low energy conformers of the 6-6 ring systems would be a good idea to ensure that
the conformation is not drastically different or require a strategic nitrogen placement to avoid major
conformational changes from the parent molecule.
Switching focus to the five-membered heterocycle substitution on the Markush structure has a lot
of possibilities as well (Fig. 1B). The claims cover a pyrazole and imidazole
in specific orientations. That leaves the possibility of regioisomers where the nitrogens are placed in
different places around the ring including N-linked to the central core. There is a possibility of
saturating the ring but this would introduce stereocenters and potentially be a more complicated synthesis.
Saturating the ring would likely have drastic effects on the conformation, although flattening out the
saturated ring in the form of a lactam or urea may help with this. Overlays of 3D conformers could
quickly answer this question. Skipping ahead to the R2 and R3 substituents,
there is a possibility of forming a fused bicyclic ring that is either saturated or unsaturated.
Bringing R2 and R3 together to form a ring is not covered in the claims.
Maybe, a 6-5 ring system would be tolerated instead of alkyl substituents. Those are just some of the possibilities
to differentiate that I have identified looking at the Markush structure; the world is your oyster! It will likely take several
strategic designs to determine a
path forward and modeling will certainly help prioritize. Now, I’ll move to the R groups and see where there are more
opportunities to differentiate from these claims.
R1 substitutions:
Looking at R1, the claims draw out five specific heterocycles that are 6-5 systems (Fig. 2A).
It’s a little odd that the claims focus on these heterocycles specifically,
but the authors could have been limited by other prior art. Determining this would require some structure searches
in a database like SciFinder or Reaxys. There are similar
possibilities as the central core. Different nitrogen substitutions are possible,
ring expansion to a 6-6 system, flipping the 6-5 system, and introducing saturation.
It is interesting that the two nitrogen bridged ring systems have a different R group representation
compared to the others (R6 and R10). R6 is much broader than R10,
which could indicate the R10 vector is limited in terms of tolerability within the binding pocket.
Another possibility is these bridged systems had worse ADME properties and the group decided to put resources elsewhere.
There are only three examples within the patent application that are not an indazole including
two with a bridged nitrogen 6-5 system and one example of the benzimidazole
(Fig. 2B). The potencies of these three exemplified molecules (Ex. 76, 86, and 94) are reduced compared to the indazole containing
molecules. The lack of other examples makes it difficult to tell if there are opportunities here for differentiation.
I would still take a shot at differentiating but could prove to be challenging. Structural information would make a big difference here to make
decisions about what to prioritize.
Subtle changes at first in this area of the molecule would probably be best initially. There is always a possibility
that other ring systems were tried and proved to be inactive.
R2 and R3 substitutions:
- R2 for B-A is -N-C= has the meaning of C1-6-alkyl-, C3-6-cycloalkyl-, C1-6-haloalkyl-;
- R2 for B-A is =C-N- has the meaning of C1-6-alkyl-, C3-6-cycloalkyl, C1-6-haloalkyl-, C1-6-alkyl-O-, HO-, H2N-, C1-6-alkyl-HN-, (C1-6-alkyl)2N-;
- R3 is H-, or C1-6-alkyl-, C3-7-cycloalkyl-, C2-6-alkenyl-, C3-7-heterocycloalkyl-, each optionally substituted with a group selected from F-, HO-, Me-, EtO-, NH2(O)C-;
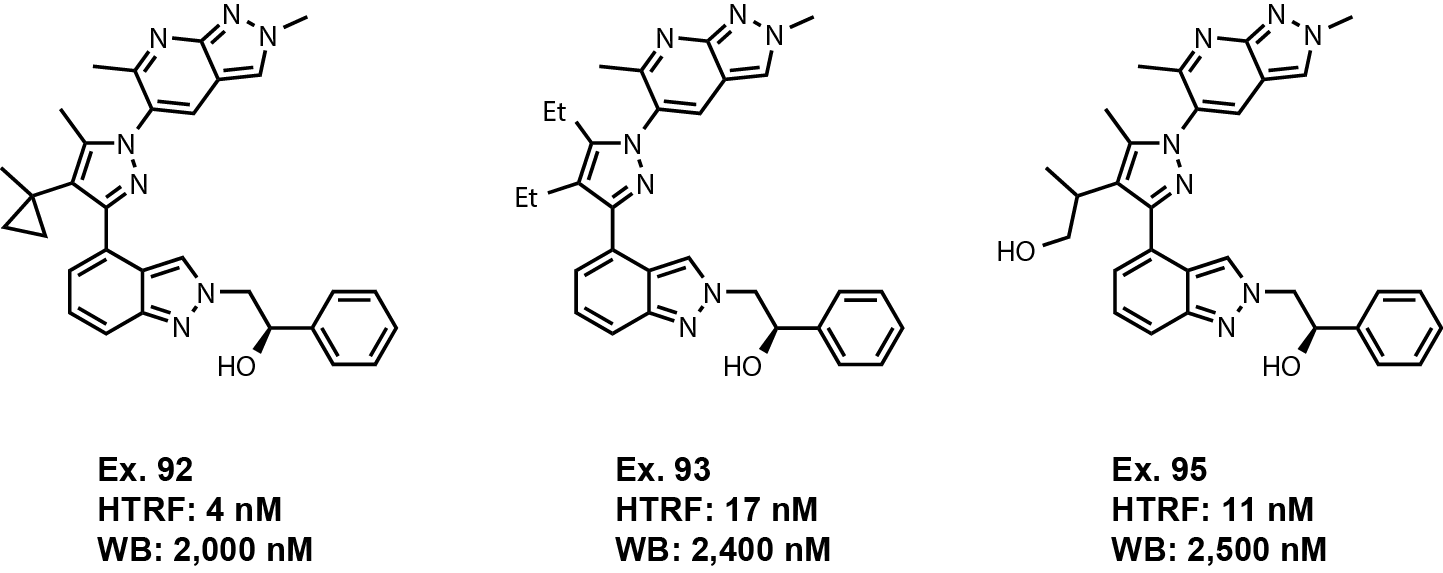
The claims surrounding R2 and R3 substitutions are quite broad considering
that the vast majority of the exemplified molecules have R2 as a methyl and R3
as an isopropyl or cyclopropyl. There are only three examples that expand beyond these substituents including
examples 92, 93, and 95 (Fig. 3). Examples 92, 93, and 95
lose 10 to 100-fold activity in the whole blood assay compared to examples with the methyl and
isopropyl/cyclopropyl at R2 and R3. But these examples are within 3-fold of the most
potent examples. This is where it is unclear if these substitutions have an actual affect on activity or if
the differences observed are due to protein binding values. I have a hard time believing these molecules have
that big of difference in protein binding to other more potent analogues, but the properties have to be evaluated
to find out exactly how these substitions are affecting activity.
Taking these examples into consideration, the claims do not cover halogens or a cyano group
for R2 and R3. Halides could be a good replacement for a methyl group and
maintain activity. If you want to get into the nuance, R3 could be a chlorine substituted
cycloalkyl group or heteroatom substitutions using O, N, or S (e.g. MeO- or N(Me)2-). Not too many
medicinal chemists would be keen on alky chlorine substituents, but it depends how desperate you are.
As mentioned above, R2 and R3 coming together to form a ring is not covered.
I like this approach and putting a cyclic group that is saturated or unsaturated here could lead to a
nice point of differentiation. If the heterocycle in the Markush structure is fully or partially
saturated, spirocycles or quaternary carbons could be tried here as well.
R4 and R4b substitutions:
- R4 is H-, F-, or HO-;
- R4b is H-, F-, Cl-, Br-, NC- or HO-;
The claims for R4 and R4b (shown on the Markush structure in Fig. 1A) are narrow.
There is only one example in the patent application outside of an unsubstituted indazole.
Example 35 has a fluorine substitution and maintains reasonable activity compared to
unsubstituted indazole examples. These claims are likely narrow because substitutions
at these positions are not tolerated, but the lack of examples make that impossible to
know without trying. For R4, it might be worth making a couple of analogues
with chlorine, methyl, or cyano substitutions. As mentioned above, introducing nitrogens
on the indazole could be a path forward as well.
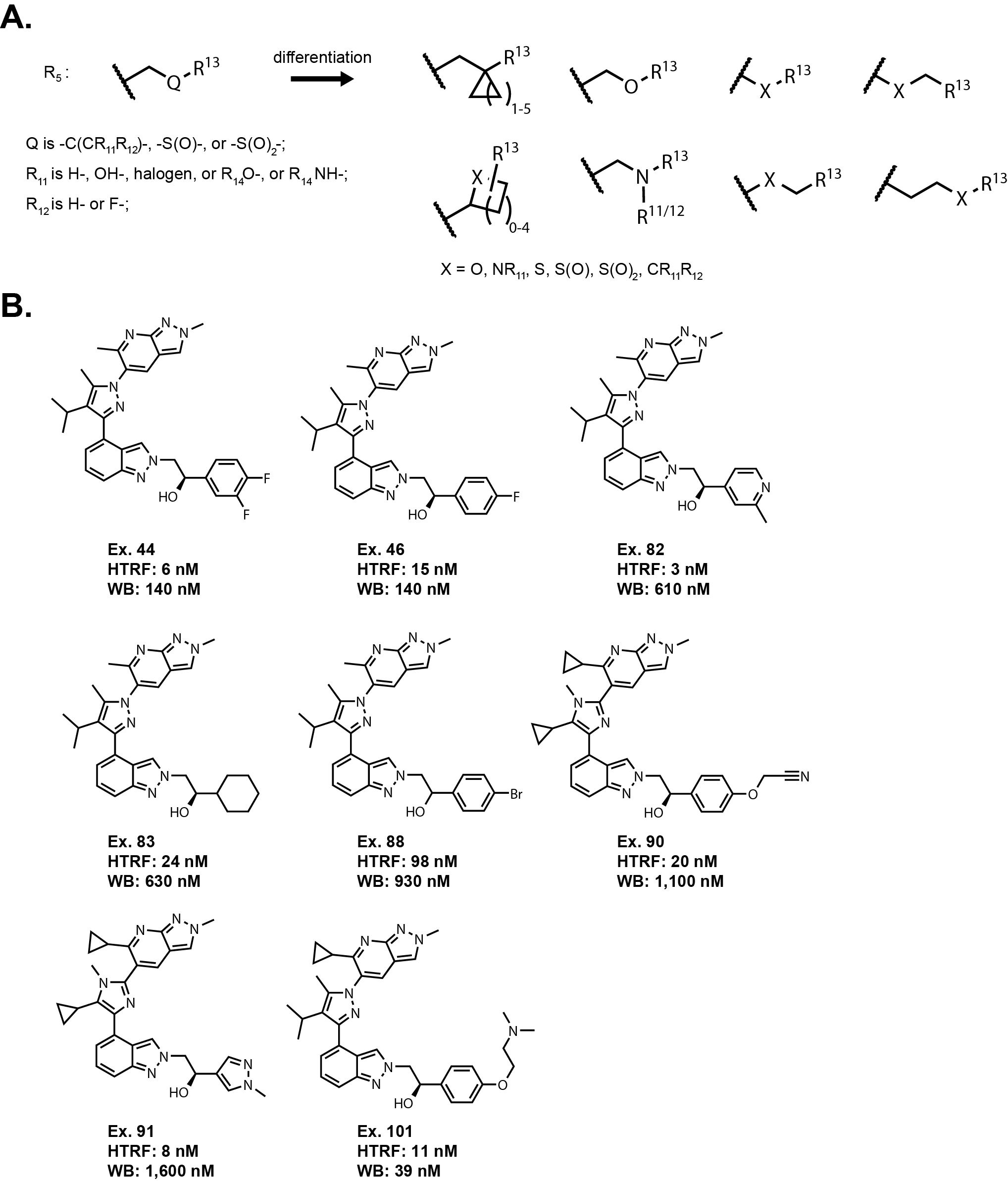
R5 Linker:
R5 extends from the indazole nitrogen as a two atom linker where the first
atom is defined as a methylene group and Q is either a carbon, sulfoxide, or sulfone.
If Q is a carbon, then there can be additional substitutions R11 and R12.
There is quite a bit that can be differentiated here starting with length of the linker.
The linker could be shortened or extended by an additional atom. The Q could be changed to
an oxygen or nitrogen. The linker itself could include a cycloalkyl or heterocycloalkyl
group. R11 and R12 could come together to form a spirocycle as well.
The claims also left out having R11 and R12 as a cyano group or
disubstituted amine, even though it covers a monosubstituted amine.
R13 is broadly defined. It appears based on the examples that
this area is sensitive to subtle changes. The predominant exemplified group is
an unsubstituted phenyl, but there are certain small substitutions such as fluorinated
phenyls examples 44 and 46 that maintain reasonable potency in the whole blood assay.
There are several other examples such as 82, 83, 88, 90, and 91 where quite a bit of
potency is lost in the whole blood assay. There is an outlier, example 101, where a 4-oxo(2-dimethylamine)ethyl
substituent off of the phenyl group maintains good potency. Since it is a protonatable amine, it could be related to
protein binding differences because putting a positive charge on a molecule can drastically affect the properties of the molecule.
Conclusion
I will stop going through the claims at this point as the rest of the R groups are quite broadly defined.
Kudos to anyone that has made it this far in this very long and likely dry blog post. Hopefully, you learned something
along the way.
I do include GAPS for the additional R groups in Table 1 but will not discuss them.
Even with these claims there are multiple points to differentiate. Some changes are higher risk
but even making small changes to the Markush indazole and heterocycle, and then making a change to the R5 linker gives three points of
differentiation. In a limited resource environment it is difficult to pursue all points of differentiation at once. That is where computational modeling
can save a lot of time and effort by deprioritizing molecules that are unlikely to have the same binding mode as the literature molecules.
A great place to start a program based on literature is to synthesize some of the most promising compounds from patent applications and
publications. It is good practice to verify reported activities and characterize physicochemical and DMPK properties. This can provide a baseline and help priortize what to pursue first. Besides making literature compounds, starting with small changes to molecules including truncating
molecules to simple pharmacophores can be a useful task. Getting an idea of what parts of the molecule impart potency will give an idea of where
differentiation will be tolerated. I will stop there. Thanks for reading.
The site does not have a comments section yet! Hopefully, very soon! Until then please drop me a line at jwiden@chemjam.com.
If you provide comments on my articles I reserve the right to post them on this website as additional commentary. My goal is to have an open discussion!