Violet Therapeutics are Targeting EphB3 for Neurodegenerative Diseases including Alzheimer's
September 2nd, 2025 By John Widen
Most research efforts towards disease modifying therapies for Alzheimer's Disease (AD) focus on lowering Aβ plaque in the brain.
The two hallmarks of AD (but not sufficient for diagnosis) are deposition of Aβ plaques and Tau neurofibrillary tangles.
Aβ plaques are produced by sequential processing of Aβ precursor protein (APP) by
two proteases, β-secretase (BACE1) and γ-secretase,
which produce two aggregation prone peptides Aβ42 and Aβ43. Aβ42 is the dominant peptide form in plaques.
The aggregation of these peptides is thought to be the root cause of neuroinflammation and neuron loss
in the brain leading to cognitive decline. Hence, lowering Aβ plaques in brains should either slow or reverse these effects.
Targeting the clearance of Aβ plaque in the brain has resulted in many
clinical failures
to slow or reverse cognitive decline in AD patients. I won’t go through all of the clinical failures over the past three decades
because that might take just as long. Currently, there is a lot of resources being put
into Aβ-clearing antibodies that recognize Aβ plaques and facilitate clearance through immune activation.
These antibodies have been controversial because clinical data has been at best confusing and largely suggests
this approach
does not make a clinically significant difference
in cognitive decline.
Lecanemab and Donanemab
were approved by the FDA in 2023 and 2024 respectively but not because they demonstrated efficacy in humans.
These antibodies do clear Aβ plaques as measured through PET or MRI imaging, but despite the reduction in plaque,
did not slow or reverse cognitive decline in a clinically meaningful way. That might be a provocative statement,
but I think it is true.
Lecanemab, Donanemab, and other Aβ-clearing antibodies are limited by a life-threatening
side effect referred to as
ARIA (Amyloid-related Imaging Abnormalities), which results from brain
swelling and hemorrhage. This side effect is relatively rare but is difficult to predict and
has led to the death of patients. Interestingly, patients that are heterozygous and homozygous
carriers of APOE4 have an
increased risk of ARIA when administered an Aβ-clearing antibody.
A new antibody developed by Roche, Trontinemab,
finished a Phase 1 trial recently (NCT04639050) and claims
that this antibody has reduced ARIA incidence. However, it still occurred in three out of 114
patients leading to one death. Whether Trontinemab reduces ARIA risk in larger cohorts will be
evaluated as this treatment progresses through clinical trials.
I do hope that Aβ-clearing antibodies result in reducing cognitive decline in AD patients.
If there is a beneficial effect from this approach, the treatments will have limitations including
the administration route, increased risk for ARIA in APOE4 carriers or patients taking certain
medications as well as cost and accessibility. New approaches to develop disease modifying
therapies to treat AD and other neurodegenerative diseases are always welcome.
That brings us to a new(ish) company called Violet Therapeutics that is doing just this.
Violet Therapeutics is a platform company that was formed based on a technology developed in
Francisco Quintana’s lab
at Brigham and Women’s Hospital and Harvard Medical School.
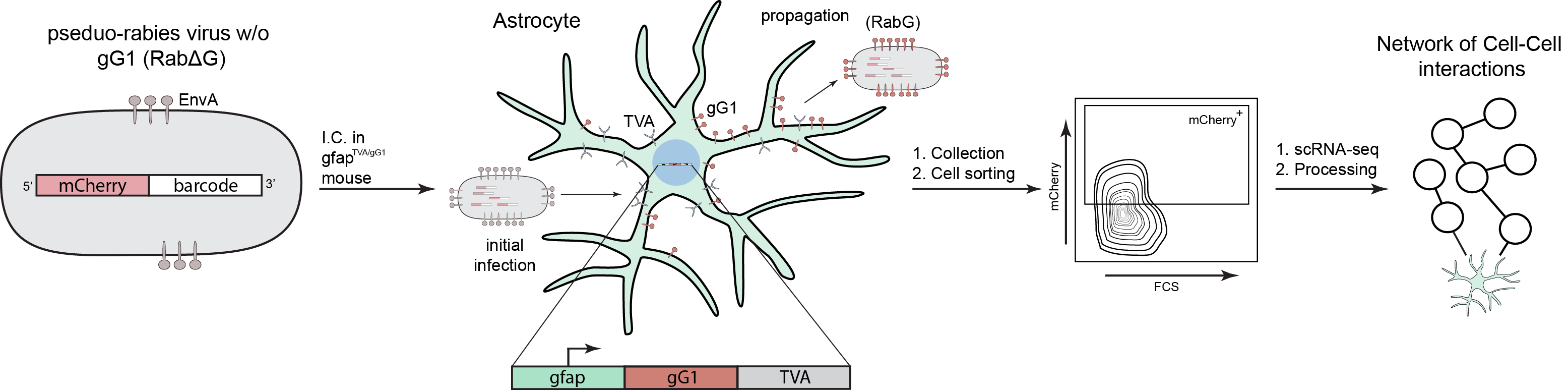
The platform combines viral barcoding and single cell RNA sequencing (scRNA-seq).
The technique is called RABID-seq
because it uses glycoprotein G–deficient pseudorabies
virus (RabΔG) to infect specific cell types with a plasmid containing a fluorescent tag
(e.g. mCherry) and several mRNA barcodes that are readable using scRNA-seq.
Violet Therapeutics is using RABID-seq as a discovery engine to identify proteins and
pathways involved in cell-cell interactions at the single cell level, which is super cool!
The cell-cell interactome screening occurs in vivo and is quite an involved process
(Fig. 1). In the publication describing RABID-seq, Quintana’s lab uses a mouse model
of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE). The experiment requires a
mouse containing glycoprotein G and EnvA receptor (TVA) under the control of a Gfap promoter
(gfapTCA/gG1). Gfap is a protein that is exclusively expressed in astrocytes and the
TVA is required for infection by the pseudorabies virus used to deliver an
mCherry-containing barcoded plasmid. Therefore, only astrocytes can express the TVA
receptor and receive the plasmid used for tracking during the initial infection.
Introducing this infection into astrocytes allows for tracking of cells that come in
contact with astrocytes because the initially infected cells go on to produce virus
containing the plasmid and Glycoprotein G, which is required for infecting other cell types.
In this study, the group compares cell interactions between EAE and naïve mice as well as
differences between high and low inflammatory responses in cells.
After providing time for cell-cell interactions to occur in vivo,
the mice are sacrificed, and their brains are processed for flow cytometry. Cells with
fluorescent signal from mCherry indicating they contain the transduced plasmid have their
mRNA sequenced. The data requires some cleanup steps that involve detection of the encoded
barcodes and building astrocyte-centric cell interaction networks. The publication focuses
on two protein pairs involved in cell-cell interactions: Sema4D-PlexinB1/B2 and Ephrin-B3-EphB3
(Eph receptor interacting proteins-Ephrin type-B receptor 3). A brief search of Sema4D-PlexinB1/2
didn’t seem to turn up any small molecule modulators but there were some reports of antibody
development towards these targets. I did find one report identifying
macrocyclic peptide inhibitors
towards plexin B1 (PlxnB1). However, these studies require brain penetrance for evaluation in animal models.
It seems that there is a gap in small molecule modulators for this protein pair. This might be the
reason the remainder of the publication focuses on the Ephrin-B3-EphB3 interaction.
EphB3 is part of a superfamily of
Eph (erythropoietin-producing hepatocellular carcinoma)
receptors that have two subclasses EphA1-10 and EphB1-6. Eph receptors and their ephrin
counterparts are involved in cell-cell interactions and have been studied as therapeutic
targets for their involvement in cancer and neurodegeneration (Fig. 2).
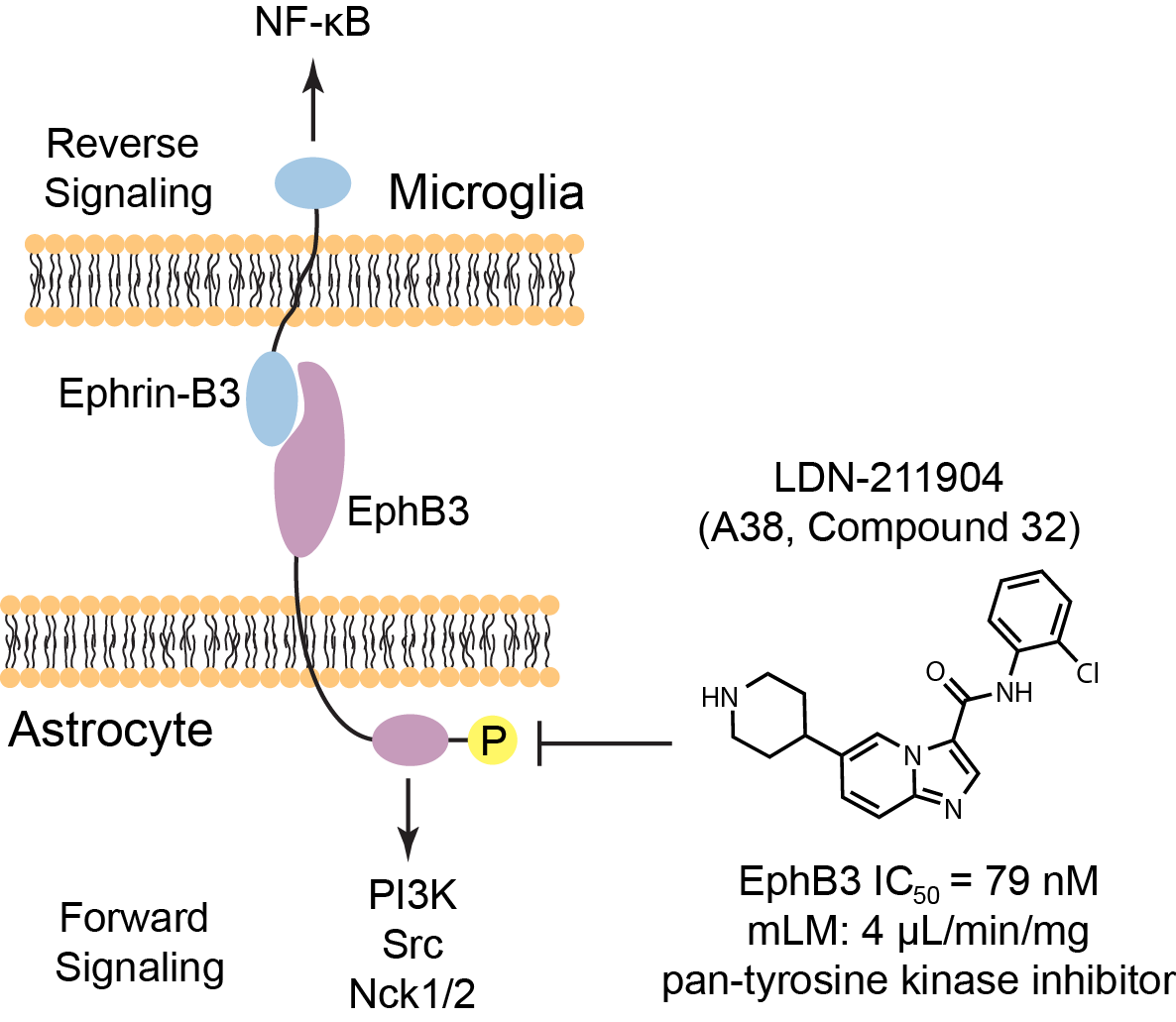
EphB3 is largely expressed in the CNS, has a receptor tyrosine kinase domain,
and is involved in axon guidance. Upon binding of an activating Ephrin,
EphB3 undergoes autophosphorylation for downstream signaling through non-catalytic region
of tyrosine kinase adaptor protein 1 (Nck1) and Nck2, phosphoinositide 3‑kinase (PI3K),
and Src family kinases. Eph receptor side signaling is referred to as forward signaling.
There is also reverse signaling through Ephrin-B3 that can activate the NF-κB pathway.
EphB3 is expressed on the surface of astrocytes but not on microglia,
whereas Eprhin-B3 is largely expressed on microglia.
Quintana et al. utilized genetic knockdown and knockout experiments in cell-based
and mouse models of multiple sclerosis to validate that the Ephrin-B3-EphB3 interaction is
involved in proinflammatory signals contributing to neuroinflammation.
The authors also used a small molecule they refer to as “A38” to demonstrate chemical
inhibition of EphB3 reduces inflammatory signaling. It is unclear where the name A38 came from,
but this molecule is reported as a tyrosine kinase inhibitor in a
2009 publication as
compound 32, LDN-211904. It is also sold at MedChemExpress under the LDN-211904 name.
The reported tyrosine kinase inhibitor LDN-211904 inhibited EphB3 kinase activity by
measuring 33P incorporation into a BTK-peptide with an IC50 of 79 nM.
The molecule also inhibited EphB3 autophosphorylation in the presence of ephrin-B3 ligand in a
stably expressing EphB3 HEK-293 cell line (>95% inhibition at 10 µM).
LDN-211904 is stable in mouse liver microsomes with reported CLint of 4 µL/min/mg.
Overall, it seems like a reasonable starting point for medicinal chemistry efforts.
It is predicted that this molecule would be brain penetrant, although this has
not been demonstrated in the public domain. However, the inhibition observed in
mouse brain by Quintana et al. supports this notion. The only major issue up to
this point is kinase selectivity. LDN-211904 was screened against 288 kinases at
5 µM and hit a variety of tyrosine kinases including other Eph receptors,
ABL, BLK, BMX, BTK, EGFR, FYN, HER4, KIT, PDGFR, SRC, YES, and others.
Unfortunately, it appears to be a pan-tyrosine kinase inhibitor.
The lack of selectivity diminishes the results in Quintana et al.
but the knockdown and knockout experiments are still very compelling for EphB3.
Violet Therapeutics also think this is so.
To address the selectivity issue, a different group at UCLA in collaboration with AstraZeneca and
Rockefeller University
took a bioinformatics approach
to identify selective covalent EphB3 inhibitors!
When I started looking into this it was a very pleasant surprise that the road led to covalent
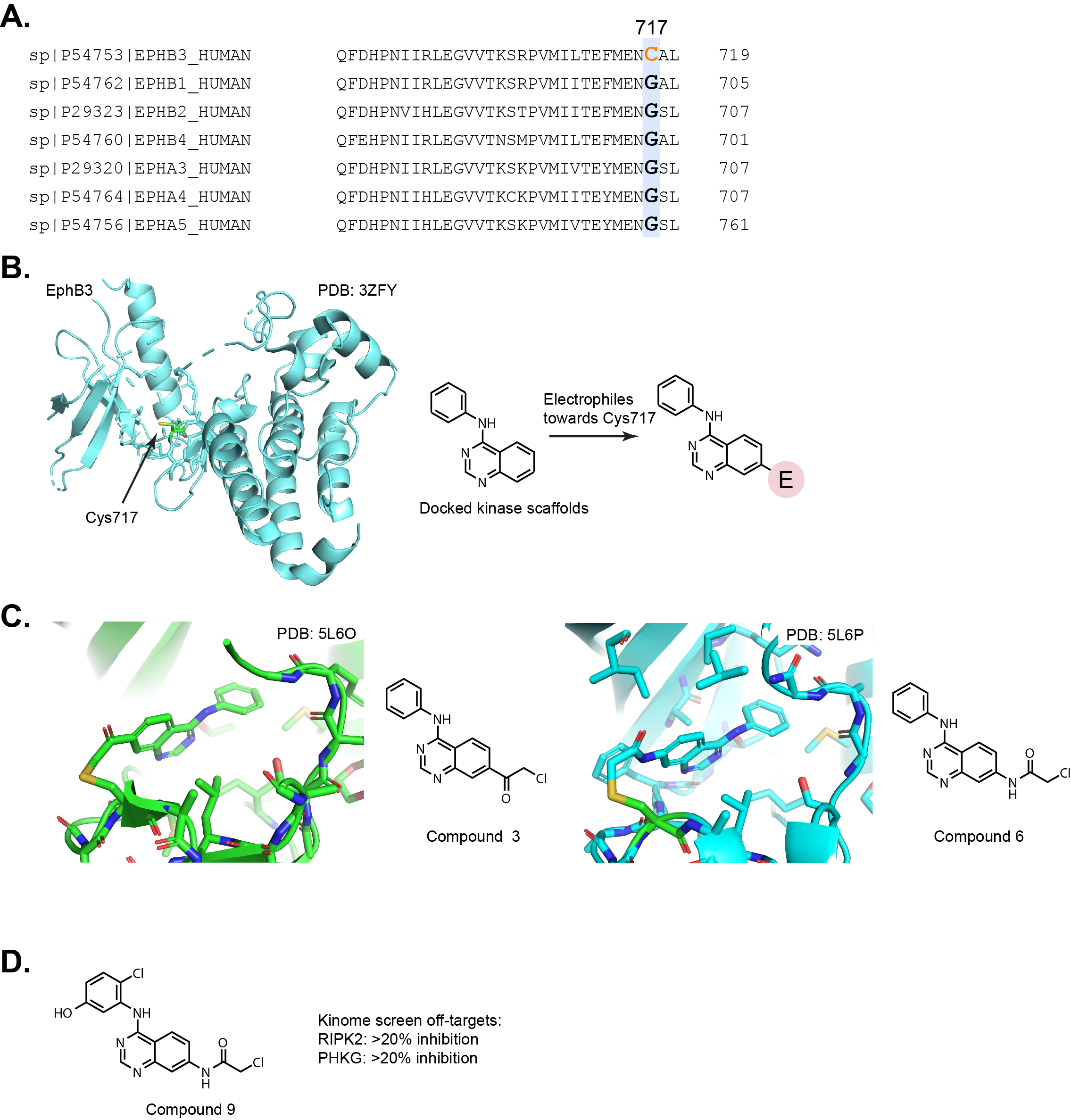
inhibition of EphB3! This group made an astute observation that EphB3 has a cysteine (Cys717)
near the hinge region of the ATP binding site. No other EphB receptors or closely related EphA
receptors have a cysteine at this position (Fig. 3A). In fact, only two other
kinases LKB1 (Liver Kinase B1, a.k.a Serine/threonine kinase 11, STK11) and PINK1 (PTEN Induced Putative Kinase 1) out of the kinome contain a similar cysteine. However, the analogous
cysteines on LKB1 and PINK1 are likely inaccessible from the ATP binding site due to
differences in the hinge region.
The group used a previously reported PBD structure (PDB: 3ZFY) to dock pharmacophores
into the EphB3 active site (Fig. 3B). This led them to focus on a quinazoline
core as the starting point to attach various electrophilic groups to target Cys717.
This approach to target cysteines is quite challenging and fails more often than not.
However, two quinazolines containing an electrophile out of seven synthesized did
inhibit EphB3 kinase activity in a time-dependent manner as determined by a washout
experiment and measuring the IC50 values at a later time point.
The three electrophiles were an α-fluoro ketone, α-chloro ketone, and α-chloro acetamide,
which are quite reactive cysteine electrophiles. I will mention here that they did not
characterize the kinact/KI, or the values separately, of the molecules
to determine the binding component or inactivation efficiency. Instead, they compared single
point IC50 values, which can be misleading when comparing irreversible inhibitors.
Nonetheless, the group demonstrated inhibition in cell-based assays and reported two X-ray
crystal structures of compound 3 and 6 bound to the active site and covalently attached to Cys717 (Fig. 3C).
Kinase selectivity for compounds 6 and 9 are reported against a panel of 98 kinases.
Compound 6 inhibited EGFR to a stronger extent than EphB3. Compound 9 inhibited only
RIPK2 and PHKG2 >20% from baseline in the screen, albeit the compound was evaluated at
50 nM at a single time point (Fig. 3D). I think additional follow-up experiments
would have to be done to demonstrate selectivity. Additionally, the group synthesized an alkyne
probe to visualize EphB3 within a cell proteome using an in-gel fluorescence labeling experiment.
The gel does show a strong band for EphB3 but there are also a variety of other dose-dependent
bands at concentrations ranging from 3-300 nM, suggesting the compound is unsurprisingly
reacting with many other cysteines within the cell proteome. Although the electrophile used
to identify inhibitors of EphB3 are reactive, this is potentially a nice starting point for
medicinal chemistry efforts and likely serves as a nice probe molecule.
This finally brings us back to Violet Therapeutics, which secured a
$10.6 million seed funding round in 2023.
EphB3 is the only drug target listed on their website and
recently gave a presentation
focused on the protein target at the AAIC (Alzheimer’s Association International Conference)
at the end of July (2025). There are no disclosed molecule structures in the slide show,
but it seems Violet Therapeutics is interested in developing EphB3 inhibitors as disease
modifying therapy for AD. They provide some DMPK and toxicity data for their tool
molecule VT-001. The molecule has a Kp,u,u of 0.4 and no off-targets in a 196
kinase panel selectivity screen, although the concentration and time of incubation is not reported.
VT-001 seems to have reasonable in vitro tox metrics with no CYPi and only one off-target
in a SafetyScreen 44; only activity observed is inhibition of AchE (acetyl choline esterase) at 3 µM,
but no % inhibition or any follow-up data is presented. They do report 37% hERG inhibition at 10 µM.
But, these metrics must be taken with a grain of salt considering their compounds are irreversible covalent.
Hopefully, this group is doing further characterization to make sure that these off target inhibitory
effects are not time-dependent.
Violet Therapeutics evaluated VT-001 in several pre-clinical rodent models including EAE,
the previously mentioned mouse model for multiple sclerosis, as well as two AD models, 5xFAD
and PS19 tauopathy mice. The tool compound rescues cognitive deficits and reduces neuroinflammation
in both pre-clinical AD models. Levels of Aβ plaque or Tau were not reported. Instead, plaque induced genes (PIG)
was measured and a cumulative score is provided. At the higher dose of VT-001 these scores are reduced indicating
a reduction in inflammatory response. The low and high doses used are not reported. As with many biotech
companies, the data presented publicly is limited and rosy to prevent giving any meaningful data to competitors
or scare away investors.
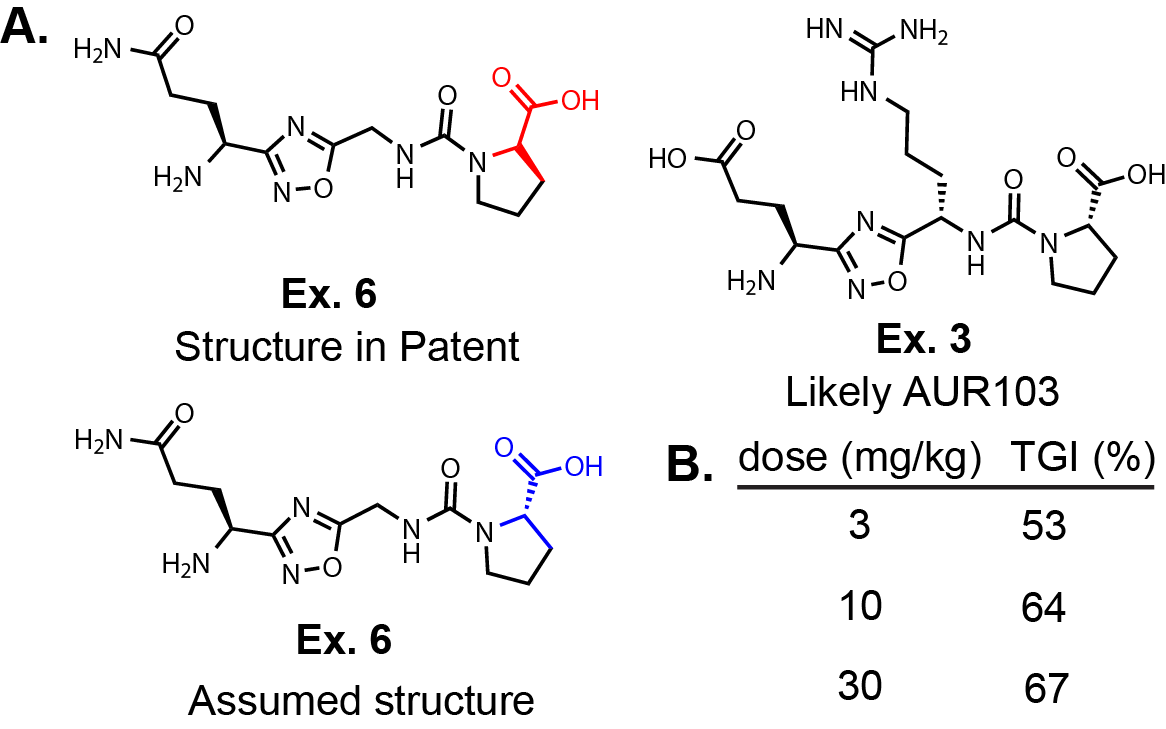
There are two patent applications published from Violet Therapeutics focused on EphB3 covalent inhibitors.
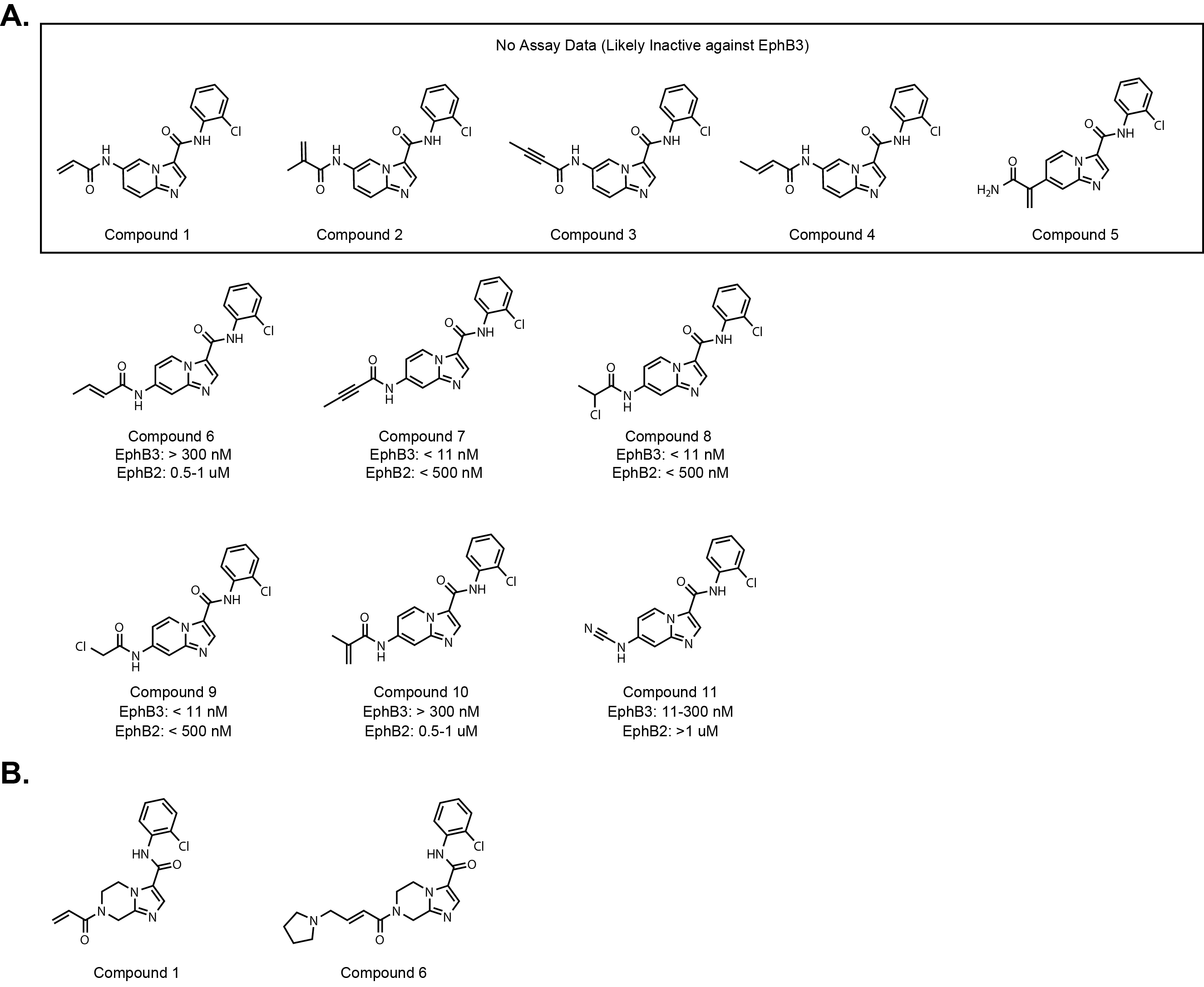
The first patent application, WO2025175088, only has 11 molecules exemplified and
are based on the LDN-211904 scaffold with an electrophile extending from the six or
seven position of the imidazopyridine (Fig. 4A.).
The only difference between the molecules is the electrophile attached.
Compounds 1-5 do not have biological data meaning they likely do not have any activity.
The most potent compounds towards EphB3 are 7, 8, and 9, which are a
butynamide, α-chloroethylamide, and α-chloromethylamide respectively.
But again, single time point IC50 values for irreversible inhibitors
can be misleading. So, there is no way to tell which compound has the best
inactivation efficiency. Interestingly, the 7-position acrylamide is not reported in
the patent application but compound 6 the β-methyl acrylamide has activity in the kinase assay.
The patent also reports EphB2 data. It appears that all of the active compounds have >20-fold
selectivity but again it is difficult to draw any strong conclusions about selectivity based on these data.
The second patent application, WO2025175247, has 667 compounds.
The core of the molecule is saturated compared to the previous application,
which changes the acrylamide position quite a bit (Fig. 4B).
This change must have made a big difference in potency and selectivity based on
the improved values reported for an in vitro kinase assay.
The patent again reports values for EphB3 and EphB2. Many analogues maintain the
same core with some exploration of the electrophile. But, they seem to hone in on
making various substitutions on the β-position of an acrylamide.
This makes sense when looking at the published structures because the acrylamide is
pointing directly out into solvent. The claims focus on two specific cores and based
on other publications a ring expansion or moving some nitrogens around the core to
differentiate could be a nice path forward to generate novel chemical matter, if one was so inclined.
I would go into more detail here, but it seems that there were many mistakes made in this large
patent application regarding structures. There are a lot of misdrawn structures in the assay data table
compared to the compound structure table after the claims. For instance, in the data table,
there are some repeat structures such as compound 1 and 667, compound 7 and 663, and many others
have this same thing happening. But when you look at the compound table with the associated name,
many of the structures are different from the ones shown in the assay table. For example,
the structure for compound 571 is different in both tables. This happens for many other compounds.
I’m not quite sure what is going on and if that would affect their patent status (I’m not a lawyer).
They have picture claims and some assay data demonstrating reasonable activity.
These mistakes are likely okay in terms of patent status, but it makes it difficult to assign activity to a given compound.
EphB3 has been a drug target for quite some time now but there is not much out
there in terms of chemical matter. These molecules target the kinase domain but I wonder
if there are other approaches to inhibiting the Ephrin-B3-EphB3 interaction.
Getting selectivity between the Eph receptors and other tyrosine kinases seems to
be the major challenge with this drug target, but the identification of a reactive
cysteine near the active site provides an opportunity to gain selectivity with covalent inhibitors.
This drug target is very reminiscent of BTK (Bruton’s Tyrosine Kinases) and the discovery of Ibrutinib.
Violet Therapeutics took a ligand-based approach to identifying novel covalent inhibitors of
EphB3 by attaching electrophiles to a scaffold from a reversible, non-covalent inhibitor reported in the literature.
This is a rare example of this approach working well. Even with structural data, it is difficult to identify
the correct placement of an electrophile to react with a nearby cysteine. More often, covalent scaffolds are
discovered from a screen and optimized with an electrophile from the beginning.
It is refreshing to see a biotech company go after a protein target not named Aβ for Alzheimer's diseases.
I hope Violet Therapeutics can take this across the finish line. It seems that EphB3 could be a target for many
diseases involving inflammation, but the data presented at AAIC suggests they are focusing on multiple sclerosis and AD.
In a resource limited environment, I would think Violet Therapeutics are evaluating molecules in these expensive
rodent experiments because these are the indications being thought of for future clinical trials. Only time will tell!
I will stop there, thanks for reading.
The site does not have a comments section yet! Hopefully, very soon! Until then please drop me a line at jwiden@chemjam.com.
If you provide comments on my articles I reserve the right to post them on this website as additional commentary. My goal is to have an open discussion!
The site does not have a comments section yet! Hopefully, very soon! Until then please drop me a line at jwiden@chemjam.com. If you provide comments on my articles I reserve the right to post them on this website as additional commentary. My goal is to have an open discussion!