Part 2: The case for CD47-SIRPα small molecule inhibition
August 8th, 2025 By John Widen
Part 1 of this blog series
covered the rationale for why developing therapeutics targeting the CD47-SIRPα interaction is a good idea.
As a brief recap, CD47 is ubiquitously
expressed on the surface of human cells as a “don’t eat me” signal. Binding of CD47 to SIRPα, which is expressed on the
cell surface of macrophages, inhibits phagocytosis.
In the context of cancer, CD47 is overexpressed on tumor cells to evade the immune system and program the
tumor microenvironment to promote tumor growth and progression. CD47 and SIRPα antibodies as well as a small
molecule modulator of CD47 have progressed into phase II clinical trials after an initial
hiccup with the
first-in-class CD47 antibody, magrolimab, which had dose-limiting toxicities. Additionally, blocking the
CD47-SIRPα interaction has been demonstrated to promote the removal of atherosclerotic plaques in pre-clinical studies
and
a small human feasibility study.
For Part 2, I’m going to cover the literature for small molecule inhibitors of the CD47-SIRPα interaction.
But, before I get into the small molecules, I wanted to mention the ongoing clinical trials for CD47
and SIRPα targeting antibodies registered with clinicaltrials.gov (Table 1). There are
fifty-one active or recently completed clinical trials evaluating twenty-five different antibodies targeting
CD47 or SIRPα. Many of the antibodies are bifunctional and target a variety of other cancer related cell
surface proteins including EGFR, PD1, PD-L1, and others. There is only one registered clinical trial
evaluating a small molecule, AUR103, developed by Aurigene.
It is a busy time for CD47 and SIRPα antibodies!
Although there are a lot of antibodies being developed to target CD47 and SIRPα, I still think there is an opportunity for
small molecule inhibitors of these targets to have a significant impact on treating cancers and atherosclerosis. I really do believe a
small molecule would have a significant advantage and differentiate from all of these antibody treatments in terms of patient compliance,
manufacturing, cost, safety, and accessibility.
And because I am a medicinal chemist, I want to focus
on the published small molecule modulators, which include linear and macrocyclic peptides! Let's go through what I found in
the literature and see if there are any good starting points for a drug discovery program.
Peptidic 1,2,4-oxadiazoles
Aurigene has published two patent applications. One is WO2019138367, and the other is a selection case that has one molecule,
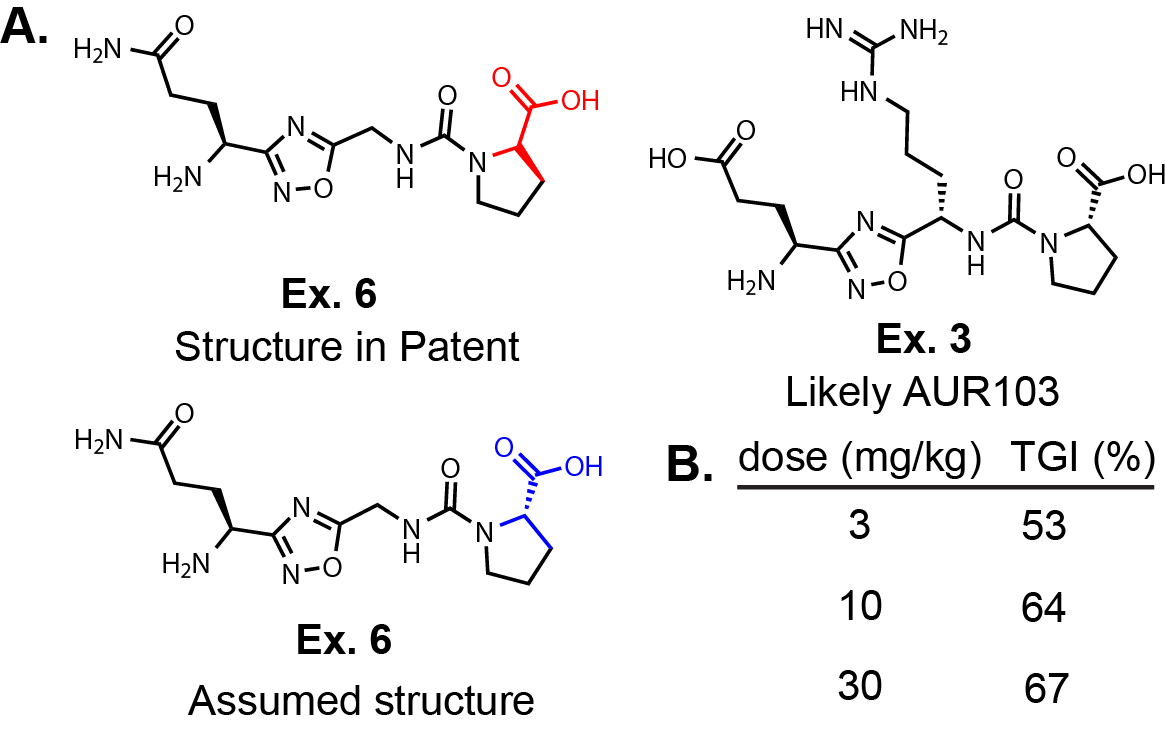
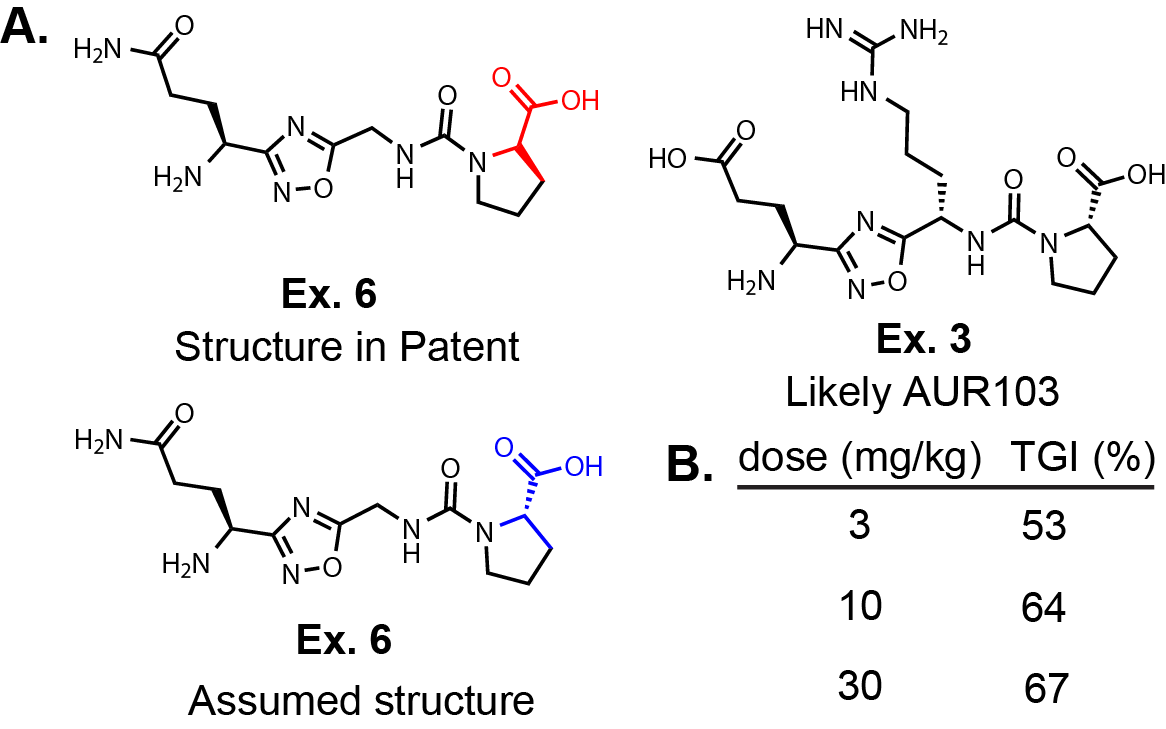
WO2025083628. In Fig. 1A, I have redrawn Ex. 6 and the only structure in the selection case
(Ex. 3 in WO2019138367). First, in WO2019138367, the stereocenter on the proline for ALL of the molecules are drawn incorrectly.
I have drawn the ‘as displayed version’ from the patent and the
likely actual structure below it. I say actual because in the synthetic methods they don’t mention D-Pro and the
selection case structure is corrected to the L-Pro. Based on their selection case, I would guess that Ex. 3 is
AUR103. Although, in the original patent application Ex. 6 is the only molecule evaluated in a syngeneic lymphoma
mouse model.
Interestingly, Ex. 6 was dosed BID P.O. for 21 days after tumors reached 75 mm2 and demonstrated
tumor growth inhibition (TGI) at 3, 10, and 30 mg/kg (Fig. 1B). There is a dose response in TGI
going from 3 to 30 mg/kg dose (3 mg/kg: 53% and 30 mg/kg: 67%), but I would expect there to be a bigger
gap between the low dose and high dose; expecting a lower response in the 3 mg/kg mice. However, it is difficult
to draw conclusions from these limited data, especially without seeing any PK data.
They do not provide any PK information or a graph showing the tumor size vs. time for the
mouse study, only these three numbers presented in Fig. 1. One possibility is that the oral exposure between
these three groups are very similar, although this is speculative. The selection invention specifically alludes to
different salt forms.
Salt forms, as well as formulation, can definitely affect bioavailability. This is particularly true when there
are protonatable amines on the molecule. Salt form, formulation, and many other factors could explain
the small difference between the low and high dose. Aurigene must have been excited about this compound because they
went ahead with a Phase I clinical trial. Godspeed. If AUR103 does demonstrate efficacy, it
will set the bar for inhibitors in the future.
Structurally, the Aurigene molecules have a central 1,2,4-oxadiazole that is formed between a single amino acid and a dipeptide.
Most of the exemplified molecules are zwitter ionic. They must be orally bioavailable based on
the route of administration in the patent application but to what extent cannot be determined.
The patent application does not cover that much chemical space, keeping the claims focused on different α-carbon substitutions. The claims also
don't expand on the 1,2,4-oxadiazole making that an easy entry point into differentiation. The examples
don't explore any non-natural amino acids either. This chemical matter is certainly an interesting starting point. I am not going to go
through the claims in anymore detail but they definitely leave a lot of opportunity to explore around the molecule. It would be a fun challenge
to go from this peptidic chemical matter to something more drug-like. To be fair to Aurigene their molecule is already entering a phase II trial!
I'm confident there is more to optimize here.
Triazoloquinazolines
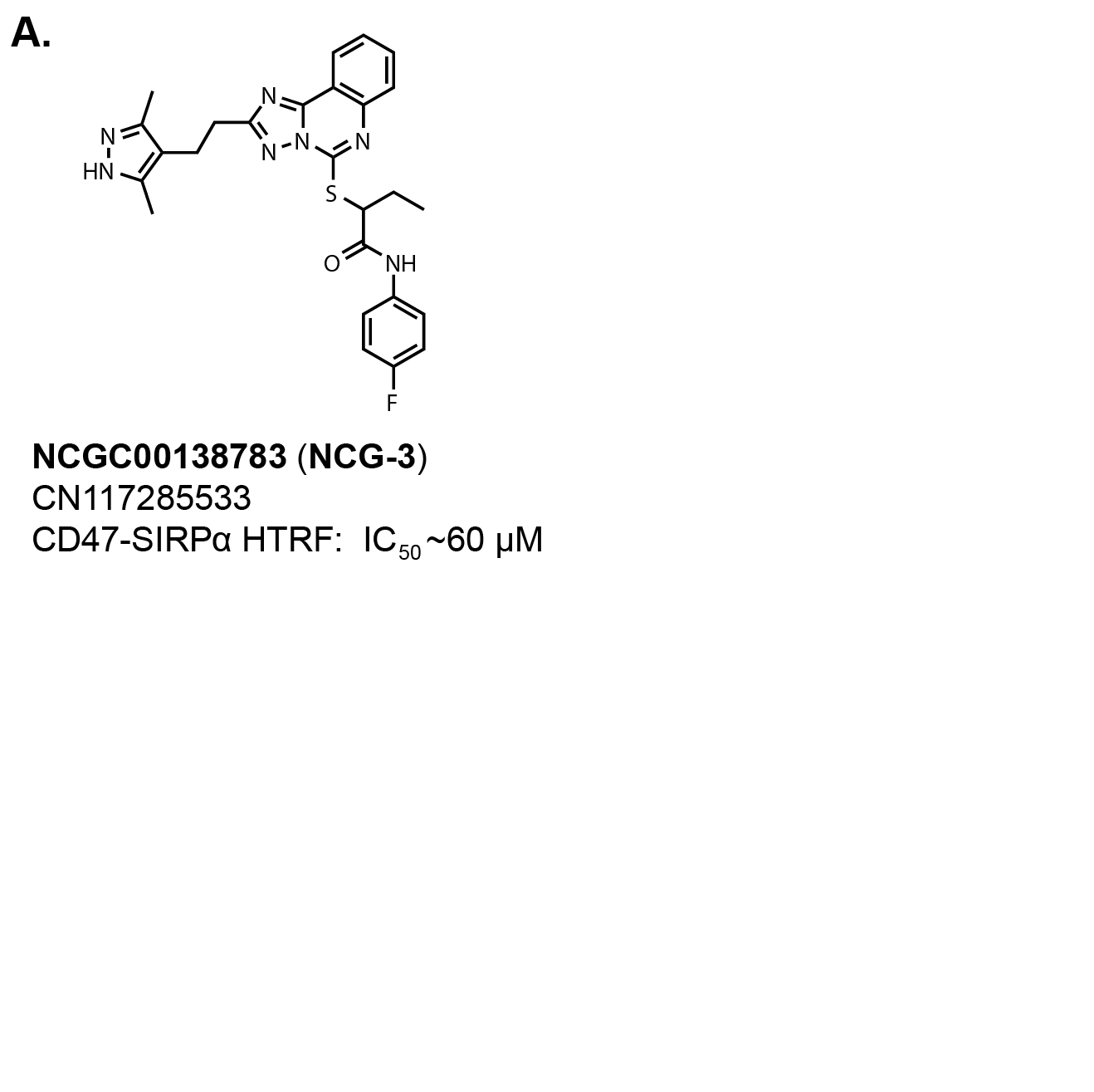
I definitely cheated with the structure-to-name function in Chemdraw for the title of this section describing the
central core of NCG00138783, which allegedly was identified from an HTS looking for inhibitors
of the CD47-SIRPα interaction described in this
PLOS One publication (Fig. 2).
The publication is odd because it exclusively focuses on assay optimization and only mentions the screening data
are deposited on PubChem (AIM: 1347059, AIM: 1347057, AIM: 1347058). There is no mention of hits or validation
anywhere in the publication.
If you search these AIM IDs on PubChem, the screening data can be downloaded in a CSV format (with SMILES for each molecule)
for viewing on medicinal chemistry software such as Vortex or
Data Warrior.
(Side note: Data Warrior is free to download and
highly recommend for aspiring medicinal chemists.) After downloading the three HTS datasets, I could not find NCG00138783
within the screening data. There really weren’t any tractable hits from the screen in my opinion, which explains the
odd publication. The compound is listed within PubChem but there is no mention of activity against CD47 or SIRPα.
There is a Chinese patent application, CN117285533, with the compound used as a positive control (NCG-3) along with
eight other molecules. The molecule is sold on MedChem Express with a reported
IC50 value of 50 µM against the CD47-SIRPα interaction. The patent application reports an IC50 ~60
µM in a CD47-SIRPα HTRF assay. All eight compounds in the patent application have the same activity.
That is a very weak affinity. I’ve already spent too many words on this molecule. So, moving on!
Quinolinones
This next set of chemical matter is more interesting. It starts with this
publication reporting
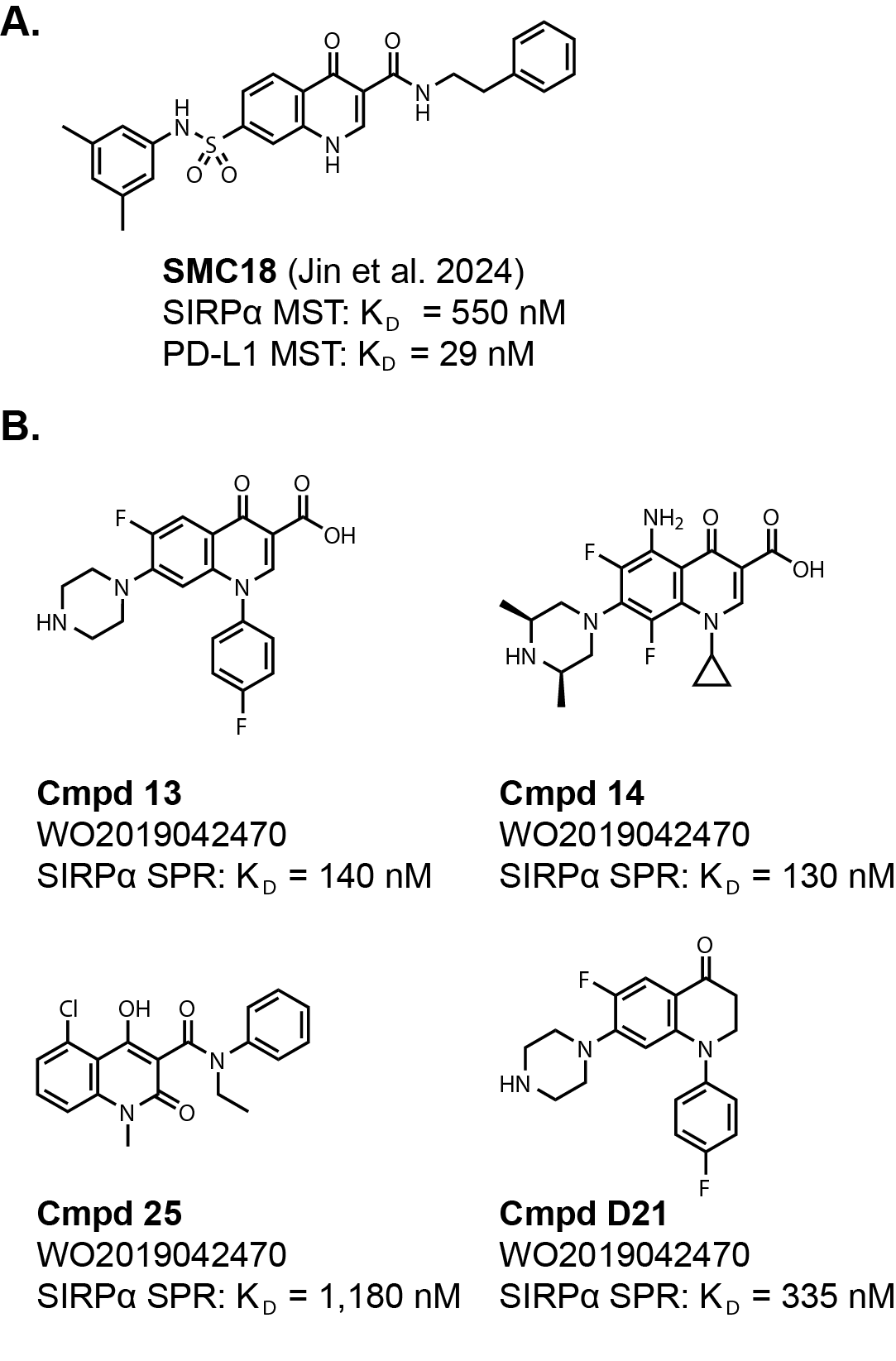
a dual CD47/SIRPα and PD-1/PD-L1 small molecule inhibitor. The star of the publication is a molecule SMC18,
which was identified from an HTS using a microscale thermophoresis (MTS) binding assay against SIRPα and PD-L1
(Fig. 3A).
The molecule has reported KD values of 550 nM for SIRPα and 29 nM for PD-L1. Pretty good binding affinity
if it can be repeated! However, the cell-based IC50 values were 18 µM for CD47/SIRPα and 20 µM for PD-1/PD-L1.
This represents a 30x and 667x shift from the binding to the cellular phagocytosis assay. This seems to be a trend for all
of the small molecules discussed thus far targeting the CD47-SIRPα interaction. The very large shifts between binding affinity
for the target and a functional assay might be concerning but I’m not familiar with the phagocytosis and cell-binding
assays. So, it might be expected. As a counter point, all of these molecules seem to have activity in tumor mouse
models despite the weak cellular activity.
There is an unrelated patent application from 2017, WO2019042470, that has similar chemical matter as SMC18
(Fig. 3B). The patent application reports KD values for the exemplified molecules using SPR.
Many of the compounds have similar affinity as SMC18 for SIRPα. It would be interesting to see if these also had affinity
against PD-L1.
This is where I’ll mention the structure. The core of these molecules is a quinolinone, which may turn some
medicinal chemists’ nose up citing likely metabolic stability and toxicity issues. Another potential issue is the
carboxylic acid substitution on the exemplified molecules in WO2019042470 that is highly represented.
Some of the compounds are zwitter ionic and it would be interesting to see how that translates
to bioavailability, elimination, and brain penetrance depending on the goals. (NOTE: A reader pointed out that
cmpd 13 is very similar to the approved antibiotic ciprofloxacin,
which IS orally bioavailable in humans!)
There is one example, D21, that has
similar binding affinity without the quinolinone core or the carboxylic acid. Likewise, SMC18 has an amide substitution
and maintains binding affinity. Additionally, the authors of the SMC18 publication report a 14-day toxicity study
in mice. Granted, SMC18 was only dosed at 2 and 6 mg/kg I.P., but at these doses the authors
report red blood cell/hemoglobin levels, ALT (alanine aminotransrerase) and AST (aspartate aminotransferase) levels, and
conducted IHC staining to demonstrate SMC18 did not have hematotoxicity and hepatotoxicity. The
route of administration for the in vivo studies is I.P. leading me to believe these compounds lack acceptable oral bioavailability
among other issues. On the otherside of things, the group very well could have never evaluated oral bioavailability.
I think this chemical series has a path forward based on these data.
It would have been nice if the toxicity study was conducted at higher doses to alleviate risk for this chemical matter.
But, even if the quinolinone had to be changed, it definitely seems possible if the preliminary SAR from the literature holds up.
I would definitely take a stab at trying to ‘scaffold hop’ and change the central core. It would be
important to determine if the massive shift from binding affinity to cell-based phagocytosis assays is relevant as well.
The in vivo efficacy data suggest it is not an issue, but definitely something to keep in mind.
This is my favorite chemical series thus far if I didn’t give that away already.
That is all for the small molecules that I could find in the literature targeting CD47 and SIRPα.
There are no data suggesting that peptidic 1,2,4-oxadiazole AUR103 binds to CD47 and not SIRPα in the public domain,
but it is listed as a CD47 targeted inhibitor on Aurigene’s website. Same goes for the weak inhibitor NCG00138783 (NCG-3).
If the peptidic 1,2,4-oxadiazole and quinolinone compounds are validated with the reported activity and direct binding to either CD47 or SIRPα,
then I think it is game on from there!
Linear and macrocyclic peptides
There are quite a few reports of linear and macrocyclic peptides as inhibitors of the CD47-SIRPα interaction.
I won’t leave my peptide people hanging and will discuss those as well. I think a macrocyclic peptide therapeutic would be a
great approach for this target! There are several relatively new companies including
Vilya,
Unnatural Products, Inc. and
Circle Pharma that focus on developing orally available
macrocyclic peptides.
There are great starting points in the literature including a set published in
Cell Chemical Biology. Select linear
and macrocyclic peptides shown in Table 2 have affinities between 3 µM to 6 nM. The asterisks next to the amino
acid or end group for the linear sequence indicate the attachment points where the macrocyclic peptides were formed. The D4 and L4
molecules used a cysteine to react with a halo acetamide at the N-terminus. The macrocyclic peptide CP5 was cyclized with a disulfide bond.
The macrocyclic peptides D4-1, D4-2, and L4-4 have high affinity (KD~10 nM) for SIRPα determined by SPR.
D4-2 also seem to have a reduced shift between affinity and the phagocytosis assay reported (KD= 11 nM vs IC50~200 nM)
compared to the small molecules mentioned above. The group also published an X-ray crystal structure of D4-2 bound to SIRPα (PDB: 2YZ1).
Whether you are interested in small molecules or a macrocyclic peptide approach to drugging the CD47-SIRPα interaction,
it seems like there are a lot of interesting starting points to get a drug discovery program started. To get started would
require synthesizing some of these molecules to verify their activity. It would be good to validate that these molecules
directly bind to CD47 or SIRPα as well as the phagocytic activity. Getting some sort of structural data should be possible
based on the abundance of various structures within the PDB database, but getting structure always involves a bit of vodou.
Definitely, worth a try though! I will stop there, thanks for reading.
The site does not have a comments section yet! Hopefully, very soon! Until then please drop me a line at jwiden@chemjam.com.
If you provide comments on my articles I reserve the right to post them on this website as additional commentary. My goal is to have an open discussion!